"Exhausted" immune cells may drive Alzheimer's

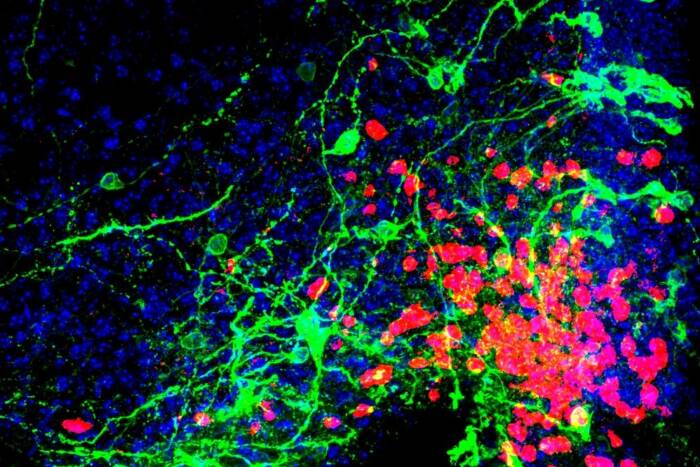
Individuals with the APOE4 gene variant are more likely to develop TIM (large blue dots) in their brain tissue (small multicolored dots), which may play a key role in driving Alzheimer’s disease.
Mice reach the twilight of their lives at around age two, the rough equivalent of 80 in human years. And when researchers introduce specific mutations into mice and then age them up, the mice can grow forgetful and irritable—eventually exhibiting signs of Alzheimer’s disease not unlike that of many elderly humans. Now, new research demonstrates that microglia, the immune cells of the brain, wither away as Alzheimer’s takes hold in both mice and humans, and that APOE4, a key gene variant implicated in Alzheimer’s, may mediate these changes.
“Older mice, and those with the APOE4 variant, have these exhausted, fatigued immune cells in their brains, and we discovered a similar phenomenon in human datasets,” says Sohail Tavazoie, the Leon Hess professor at Rockefeller. The team dubbed this new class of exhausted cells TIM, for terminally inflammatory microglia. TIM have lost the ability to efficiently remove plaque from the brain and thus may contribute to Alzheimer’s.
The study also sheds light on how the Alzheimer’s drug aducanumab may be interacting with immune cells in the brain. “When mice with the APOE4 variant were treated with aducanumab, we found that their TIM regain some functionality,” says Alon Millet, a graduate fellow in the Tavazoie lab.
Age and inflammation
Humans carry one of three variants of the APOE gene: APOE2, APOE3, and APOE4. Prior work from the Tavazoie lab has demonstrated that these variants can play pivotal roles in how the body responds to diseases from cancer to COVID, but the link between Alzheimer’s disease and APOE4 is particularly well-established: the APOE4 variant, carried by about 20 percent of the population, is considered one of the strongest genetic risk factors for Alzheimer’s.
Tavazoie, Millet, and Jose Ledo (now a faculty member at Medical University of South Carolina) spent four years developing Alzheimer’s disease mouse models that express human APOE variants and then aging them up to get a better idea of how APOE4 influences their brains as Alzheimer’s takes hold. “Generating these mice systematically was a major undertaking,” Tavazoie says. “It was an ongoing project made possible by Jose’s and Alon’s specific expertise coming together.”
The team then built a single-cell atlas of brain immune cells in these mice and identified a population of microglia riddled with signs of stress and inflammation that had not been described previously.
The brains of mice with APOE4 were overrun by TIM, while other variants had comparatively fewer TIM. And once they knew what to look for, the team also began finding TIM in human brain tissue donated by patients who had the APOE4 variant. The results suggest that APOE4 may increase the risk of Alzheimer’s by wearing down the brain’s immune cells.
The researchers also found that treating mice with the recently approved Alzheimer’s drug aducanumab improved their condition and rehabilitated damaged TIM. Interestingly, the effects of the drug were far more pronounced in mice with APOE4. And while such preliminary findings cannot be translated immediately to the clinic, “this may be a first hint that aducanumab works differently with different genotypes,” Millet says. “It’s something that clinicians should look into.”
Helping the immune system help itself
Some researchers suspect that a healthy immune system clears plaque before it accumulates in the brain, and that Alzheimer’s is what happens when that system fails, and plaque piles up. According to this theory, rehabilitating microglia too tired to do their jobs may give the brain the boost it needs to protect itself. If so, TIM would be a promising therapeutic target.
“TIM marinate in this inflammatory milieu for years, until they’re no longer able to cope,” Millet says. “If we can revert them back to a healthy state, perhaps the immune system would be able to keep Alzheimer’s in check.”
On that front, the team will now be exploring the signaling molecules that lead to TIM formation, with an eye toward contributing to the development of drugs that interfere with the process, keep microglia healthy, and reduce cognitive decline. In the long-term, this could lead to a novel Alzheimer’s therapy.
The team will also be investigating whether TIM exist in other diseases. Millet suspects that, although TIM may have escaped notice until now, these exhausted immune cells may also be involved in other diseases of the brain, from tumors to Parkinson’s. “Inflammation causes TIM to accumulate, so perhaps what we’re seeing isn’t specific to Alzheimer’s,” he says.
“Most microglia could end up as TIMs, if we give them enough time.”