Unlocking how the new Alzheimer’s drug lecanemab works
Approved by the FDA earlier this year, the Alzheimer’s therapy lecanemab is an antibody that reduces the buildup in the brain of a sticky peptide called amyloid-beta (Aβ), which is thought to be a main driver of the disease. Dispensed in bimonthly infusions, lecanemab specifically targets protofibrils, the type of Aβ clumps thought to be the most neurotoxic. Studies have shown that lecanemab slows cognitive and functional decline in early-stage Alzheimer’s patients.
But as with many other cutting-edge therapies, we know more about lecanemab’s effectiveness than we do about how it actually works. A new study published in PNAS by researchers from The Rockefeller University identifies one possible means for its impact on patients.
“We believe we’ve found a mechanism that is one of the reasons lecanemab works,” says co-senior author Erin Norris, a research associate professor in the Patricia and John Rosenwald Laboratory of Neurobiology and Genetics at Rockefeller.
Drug promises—and failures
Alzheimer’s is the most common cause of dementia in the world. In the U.S., six million people are living with the disease; 1 in 5 women and 1 in 10 men over 45 are at risk of developing it. Since the turn of the century, deaths from Alzheimer’s have increased 145%, accounting for 1 in 3 senior deaths.
The disease is dismayingly complex. Most cases of Alzheimer’s do not have a simple genetic cause, but are due to each person’s unique mix of genetics, lifestyle, environmental exposure, and risk factors, which researchers have yet to tease apart.
The slow progress on drug therapies has been equally dismaying. Until lecanemab, the most promising candidate was aducanumab, an antibody therapy that also reduces Aβ clusters from the brain. The drug gained FDA approval in 2021, but aducanumab causes devastating side effects for some patients. The worst are amyloid-related imaging abnormalities, or ARIA, which take two forms: edema, the accumulation of water in tissues, or hemorrhage. Aducanumab causes ARIA in about 35% of patients. Two other anti-Aβ therapies still in clinical trials haven’t fared much better: gantenerumab causes ARIA in 30% of patients and donanemab in 27%.
Lecanemab, however, causes ARIA in only 10% of patients. Intrigued by this low number, the Rockefeller team had two questions: why is it effective, and why does it cause so much less ARIA?
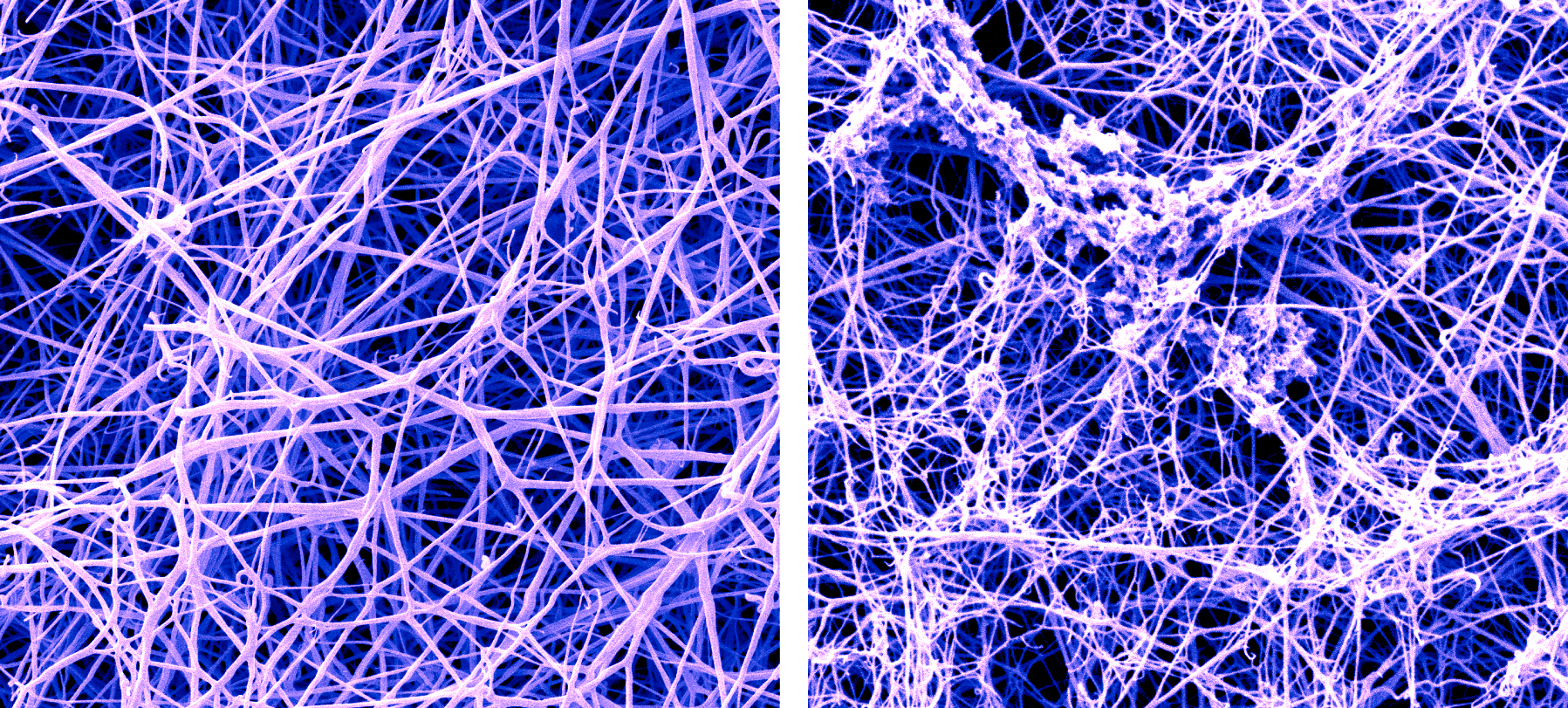
A false-color SEM image of a fibrin blood clot before and after the buildup of amyloid-beta proteins called protofibrils. (Photo credit: Anurag Sharma, Ph.D., of Rockefeller’s Electron Microscopy Resource Center, and Pradeep Singh, Ph.D.)
The Goldilocks
The researchers knew where they wanted to look: at the specific type of Aβ clumps lecanemab targets, known as protofibrils. They already knew that Aβ is responsible for activating the plasma contact system—a sort of backup to the body’s primary coagulation system, which keeps our blood from spilling out at the site of a wound—and suspected that Aβ protofibrils had a unique relationship with the contact system.
The lab had previously documented that the contact system was more activated in Alzheimer’s patients than in people without dementia, and had identified connections between the cognitive decline characteristic of Alzheimer’s and associated cerebrovascular abnormalities such as hemorrhage, decreased cerebral blood flow, and small vessel disease.
The fact that lecanemab targets Aβ protofibrils and is effective in Alzheimer’s gave them a choice opportunity. Using about a dozen different assays, studies, and analyses, the team studied the plasma from healthy donors to parse the mechanistic details. They discovered that of the multiple forms of Aβ, protofibrils were the only form that activated the contact system. It’s sort of the Goldilocks of Aβ: smaller forms were too tiny for contact system activation, and the larger form was too big—but the protofibril was just right.
Protofibrils have just the right amount of real estate for two plasma proteins, coagulation factor XII and high molecular weight kininogen (HK), to bind near each other on their surfaces. When that happens, HK is cleaved, producing a peptide called bradykinin that dilates blood vessels. Bradykinin has a variety of purposes in the body, and it can be especially useful for lowering blood pressure. But the overproduction of bradykinin leads to inflammation and edema—the ARIA side effect that has been the downfall of the other anti-Aβ therapies.
Lecanemab, they believe, interferes with Aβ in two ways: it reduces the amount of buildup in the brain and blocks protofibrils from activating the contact system, which in turn inhibits the production of bradykinin. That may explain why lecanemab causes less ARIA.
“It’s pretty amazing,” Norris says, “that the form of Aβ that specifically activates the contact system is the same form of Aβ that lecanemab targets.”
A new therapy on the horizon
The Rockefeller team is especially intrigued by this discovery because it parallels their own work on an antibody they developed called 3E8. Like lecanemab, 3E8 blocks the contact system from being activated, but it does so by targeting HK, preventing its cleavage as well as the production of bradykinin.
“If you block the contact system, you’re going to get less Alzheimer’s pathology,” says co-senior author Sidney Strickland, Rockefeller’s Zachary and Elizabeth M. Fisher Professor in Alzheimer’s and Neurodegenerative Disease.
This anti-HK therapy could be used to treat Alzheimer’s patients either on its own or in conjunction with existing anti-Aβ medications, Norris says. “You could use it as a complement to lecanemab or donanemab. With those drugs, you’re blocking Aβ, but you still get some contact system activation caused by other factors, leading to ARIA. If you block the contact system entirely, you might just do away with ARIA.”
Based on trials in primates, Strickland envisions 3E8 being dispensed once a month, potentially at the same time as one of the bimonthly anti-Aβ therapies. “So you could administer it by itself, piggyback it on lecanemab, or eventually even make a mixture,” Strickland says.
Moreover, Norris adds, if 3E8 proves effective in clinical testing—which is still far down the line—it could potentially be used to treat a variety of diseases with one thing in common: contact system activation. “The dysregulation of the contact system is involved in COVID, sickle cell anemia, hereditary angioedema, inflammatory bowel disease, sepsis, lupus, arthritis, and even cancer metastasis,” she says.




