A new class of antivirals could help prevent future pandemics
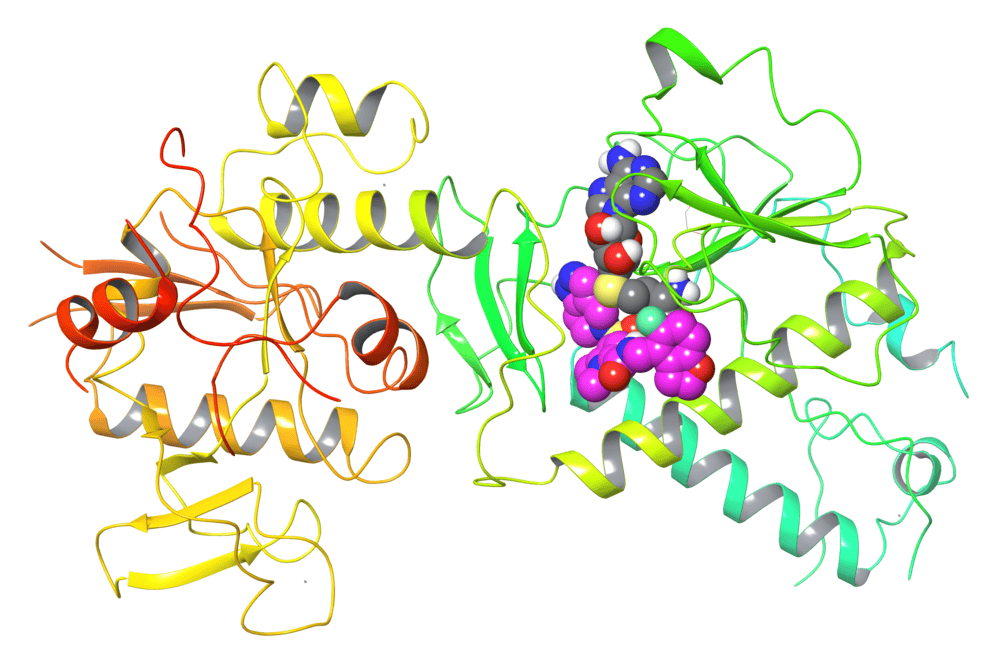
Drug (purple) that inhibits viral mRNA cap methyltransferase NSP14 for antiviral therapy. Drug binding requires the presence of SAH (grey), which is the reaction product after methyl transfer from SAM. (Credit: Tuschl lab)
The arrival of Paxlovid in December 2021 marked another turning point in the COVID-19 pandemic—an effective antiviral that has since successfully treated millions. But like many antivirals before it, scientists know that at some point, Paxlovid is likely to lose some efficacy due to drug resistance. Researchers working to stay ahead of such emerging threats have now identified a wholly new way to treat SARS-CoV-2 infections—work that may have even broader implications.
In fact, a new study by the Tuschl laboratory introduces a proof-of-concept for a novel class of antivirals that would target a type of enzyme essential not just to SARS, but also many RNA viruses, including Ebola and dengue, as well as cytosolic-replicating DNA viruses, including Pox viruses. The findings, published in Nature, may pave the way for a faster and more robust response to future pandemics.
“Nobody has found a way to inhibit this enzyme before,” says Thomas Tuschl, the F. M. Al Akl and Margaret Al Akl professor at Rockefeller. “Our work establishes cap methyl transferase enzymes as therapeutic targets and opens the door to many more antiviral developments against pathogens that until now we’ve had only limited tools to fight.”
A new way forward
The way so many RNA viruses thrive is by modifying their RNA caps, specialized structures that stabilize viral RNA, enhance its translation, and mimic host mRNA to evade immune defenses. RNA capping relies on enzymes called methyltransferases—making it a tempting target for antiviral therapies.
But most antivirals, including Paxlovid, instead focus on disrupting proteases, a different class of viral enzymes that break down proteins—largely because those enzymes were previously targeted and prevented viral spread. “Inhibiting methyltransferase required using a non-conventional RNA substrate adding a new challenge to drug to discovery,” Tuschl says.
For Tuschl, an RNA expert whose work has already led to multiple RNA therapeutics for treatment of genetic disorders, that was not much of a complication. And after he restructured his lab during the pandemic to focus on antiviral drug discovery, Tuschl realized there were clear advantages to looking beyond protease inhibitors. Tuschl suspected that viruses would be less likely to dodge a combination therapy that targeted two unrelated viral enzymes at once, such as a protease inhibitor alongside a methyltransferase inhibitor. He also realized that drugs targeting viral methyltransferase distinct in structure from the human enzyme will be highly selective and not impair human enzyme function.
In search of a molecule capable of inhibiting the SARS-CoV-2 methyltransferase NSP14, his team screened 430,000 compounds early in the pandemic in the university’s Fisher Drug Discovery Resource Center and discovered a small number of compounds that inhibited the viral cap methyltransferase NSP14, a multifunctional enzyme with methyltransferase activity.
Those compounds then went through an extensive chemical developmental process to create optimized drug candidates in partnership with the Sanders Tri-Institutional Therapeutics Discovery Institute. Compounds with improved biochemical inhibition were then subjected to cell-based assays conducted by researchers led by Charles M. Rice, who heads the Laboratory of Virology and Infectious Disease at Rockefeller. Finally, colleagues at the Center for Discovery and Innovation in New Jersey then tested the compound in mice under BL3 safety conditions and demonstrated that it could treat COVID-19 on par with Paxlovid. Tuschl and colleagues also demonstrated that the treatment remained effective even if the virus mutated in response to it, and that there was synergy when combined with protease inhibitors.
“Even in isolation, a virus would have trouble escaping this compound,” Tuschl says. “But as a combined therapy along with a protease inhibitor—escape would be almost impossible.”
Back to basics
The findings not only validate viral methyltransferases as promising therapeutic target, but also suggest that Tuschl’s particular inhibitor would have minimal side effects. “The mechanism by which the drug acts is unique,” he notes. In fact, the compound takes advantage of the unique structural features of the viral methyltransferase also requiring the presence of the reaction product of the methyl donor SAM, meaning that the lab’s compound selectively targets the virus without disrupting human processes.
“We’re not ready to test the compound in humans,” Tuschl cautions. An ideal clinical candidate needs improved stability, bioavailability, and a series of other pharmacologic properties that remain to be optimized in the long-term. “We’re an academic lab. For that, we’d need an industry partner.”
In the immediate future, the Tuschl lab is expanding this work to explore inhibitors for RSV, flaviviruses, such as dengue and Zika, as well as mpox and even fungal infections, which all share a similar enzymatic vulnerability. “This work opens the door to targeting many pathogens,” he says. “It’s a new opportunity to prepare for future pandemics.”



