Mouse Model of Diabetes Shows Synergy of Multiple Genes in Failure of Insulin-Secreting Cells
Potential Drug Target for Treatment of Type 2 Diabetes Identified

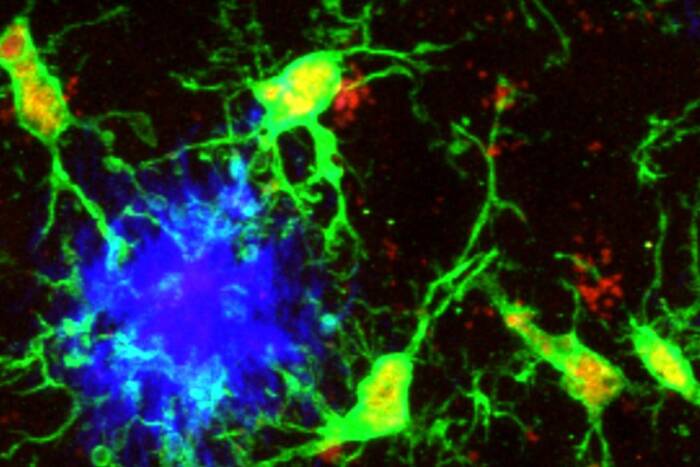
In normal mice (left), insulin-producing beta cells (red) are contained within glucagon-producing alpha cells (green), which line the periphery of islet cells in the pancreas. This architecture is disrupted in mice with mutations in the Hnf-1alpha and Pdx-1 genes (right), and impairs insulin production. This change in cell morphology also is seen in mice with mutations in Hnf-3beta and Pdx-1.
Researchers at The Rockefeller University and the Vanderbilt Medical Center have shown for the first time in a mouse model that genes predisposing for Type 2 diabetes interact in a hierarchical manner and have identified a potential drug target from these genes.
Reporting in the March 19 issue of Proceedings of the National Academy of Sciences, the researchers, led by Rockefeller professor Markus Stoffel, M.D., developed a mouse model for Type 2 diabetes that has defects only in pancreatic beta cells, the cells responsible for the controlled release of insulin.
“We have shown in a whole animal system that single gene mutations, which by themselves may not be sufficient to induce diabetes, can combine to cause severe diabetes in these animals,” says Stoffel, head of the Laboratory of Metabolic Diseases at Rockefeller.
“We also show that this effect is mediated mainly through a previously identified gene, Pdx-1, which lies at the bottom of the hierarchy, providing a possible target for improved diabetes therapies,” adds Stoffel, who is the Robert and Harriet Heilbrunn Professor at Rockefeller University.
Type 2 diabetes, the most common form of diabetes, is polygenic, meaning that more that one gene is involved in the disease. In people with Type 2 diabetes, levels of a blood sugar called glucose are too high as a result of ineffective insulin secretion or improper insulin action on target tissues such as muscle. The result is a buildup of glucose in the blood. This excess glucose is responsible for most of the complications of diabetes, including blindness, kidney failure, heart disease and stroke.
Stoffel and his colleagues have spent the last several years defining the mechanisms behind pancreatic beta cell failure. For the research published in the PNAS paper, Stoffel focused on three genes – Hnf-1alpha, Hnf-3beta and Pdx-1 – known to be involved in the development of Type 2 diabetes.
The proteins produced by Hnf-1alpha and Hnf-3beta belong to a class of molecules called transcription factors, molecules that switch other genes on or off, and are known to regulate gene activity or expression in the liver, kidney and intestine. Pdx-1 is a transcription factor responsible primarily for development of several key cells in the pancreas, including beta cells.
“Together, these transcription factors regulate a set of genes that are very important for insulin secretion,” says Stoffel. “So far, the evidence has been more or less biochemical and through cell culture systems, but it really hasn’t been proven in a whole animal.”
To test this hypothesis in an animal model, Stoffel and his research colleagues generated a polygenic diabetes mouse model with defects only in the pancreatic beta cells. These mice had mutations in the Pdx-1 gene and either Hnf-3beta or Hnf-1alpha.
Mice with a mutation in the Pdx-1 gene develop a mild form of diabetes, whereas mice with a mutation in Hnf-3beta or Hnf-1alpha genes are not diabetic. However, diabetes profoundly worsened in the polygenic mice.
The researchers found that mice born with mutations in the Pdx-1 and Hnf-3beta genes have a mild form of diabetes, like the mice with a Pdx-1 gene mutation. However, diabetes worsened after the mice were three weeks old and well into adulthood, suggesting that a combination of mutations is important for developing diabetes later in life.
“These data show that two mutations in the beta cell, one causing mild diabetes and one not causing any diabetes, can combine and produce a more severe disease,” says Stoffel.
Mice with combined mutations of Hnf-1alpha and Pdx-1 developed severe diabetes, with higher levels of glucose, soon after being weaned from their mothers, suggesting that this combination of mutations contributes to an earlier onset of diabetes.
Because Pdx-1 lies “downstream” of Hnf-3beta and Hnf-1alpha, Stoffel suggests that this gene may provide a suitable target for the development of new diabetes therapies.
“The major attraction of Pdx-1 is that its activity is restricted to the pancreas,” says Stoffel. “Hnf-3beta and Hnf-1alpha also regulate functions in parts of the body other than the pancreas. Any reduction of the activity of these two genes most likely will have adverse effects.”
Stoffel and his colleagues also examined secretion of insulin in response to injections of glucose in the mice.
In mice with no mutations, insulin secretion typically occurs in two stages. Within the first five minutes following glucose injections, there is an immediate spike in insulin secretion, produced by cells called secretory granules that sit just below the plasma membrane (the external membrane) of the pancreatic beta cell. Because this release of insulin is sometimes not sufficient to drop the glucose level, the pancreatic beta cell produces additional secretory granules during the second stage.
The researchers found alterations during both stages of insulin secretion in the polygenic mice. There was no initial spike, and insulin secretion was decreased during the second stage.
To explain these changes, the researchers studied the architecture of the pancreatic islet cells of the various mutant mice. Islet cells in mice with no mutations or only single mutations resembled those of normal mice: most of the cells in the center of the islet are beta cells; “alpha” cells, which contain the molecule glucagon (which is secreted during starvation), reside along the periphery.
However, in the islets of the polygenic mice, alpha cells were scattered throughout.
The researchers attribute this to decreased activity of a gene called E-cadherin, which produces a protein whose function is to “glue” these cells to the perimeter of the islet.
Stoffel’s co-authors are David Q. Shih, Markus Heimesaat, M.D., and Satoru Kuwajima, M.D., Ph.D., at Rockefeller, and Roland Stein, Ph.D., and Christopher V.E. Wright, D.Phil., at Vanderbilt.
This research was supported by grants from the National Institutes of Health, the Medical Scientists Training Program, the Juvenile Diabetes Foundation International, Deutsche Forschungsgemeinschaft (German Research Society) and the Human Science Frontier Organization.