Researchers Find Key to Tuberculosis Persistence in the Body
Discovery provides hints for foiling world’s No. 1 infectious killer
The tuberculosis bacterium requires a specific enzyme to cause persistent infection, a consortium of researchers at Rockefeller University and three other institutions have found. The discovery suggests that targeting the enzyme could improve therapies for TB, which claims more lives each year than any other bacterial infection.
The finding, published in the Aug. 17 issue of the journal Nature, resulted from a multidisciplinary effort by investigators at Rockefeller, Albert Einstein College of Medicine, Washington University in St. Louis and Texas A & M University, each of whom focused on different aspects of the problem.
The enzyme produced by the bacterium, isocitrate lyase (ICL), allows the TB microbe Mycobacterium tuberculosis to use fatty acids as a source of energy. Fatty acids are the most abundant source of stored energy in the body’s cells. The new findings suggest that these reserves may be exploited by “intracellular” pathogens like M. tuberculosis, which make their living by parasitizing the cells of the host. The researchers have shown in a mouse model of TB that disabling the gene for ICL in M. tuberculosiscrippled the bacterium in the disease’s later, persistent phase.

John McKinney , Ph.D.
“If we can block an enzyme that allows M. tuberculosis to persist, that might provide an avenue for attacking the bug in its latent phase,” says lead author John McKinney, Ph.D., assistant professor and head of Rockefeller’s Laboratory of Infection Biology. “A drug that targeted persistence would be quite different from conventional TB drugs, which target processes required for bacterial growth. ICL is not present in humans, so blocking its activity should not cause harmful side effects in the host.”
When M. tuberculosis infects the body, the immune system rallies to fight the invader but is unable to eradicate the microbe completely. A stalemate is achieved in which the bacteria continue to live in infected tissue, although their multiplication is inhibited. The infection can persist in a latent or chronic state for the lifetime of the host and can flare up if the immune system is later weakened.
The bug’s persistence in the body has been the main barrier to clearing the TB pathogen with drugs. Current treatments require three to five drugs for at least six months, a regimen many patients are unable or unwilling to follow. The drugs can have unpleasant side effects and in some cases cannot be taken because they interfere with other medications. Consequently, most patients fail to complete therapy unless their compliance is closely monitored.
“A drug that inhibited ICL activity or synthesis might allow us to shorten the duration of chemotherapy from six months or more to just a few weeks,” McKinney says. “That would have an enormous impact on patient compliance and cure rates.”
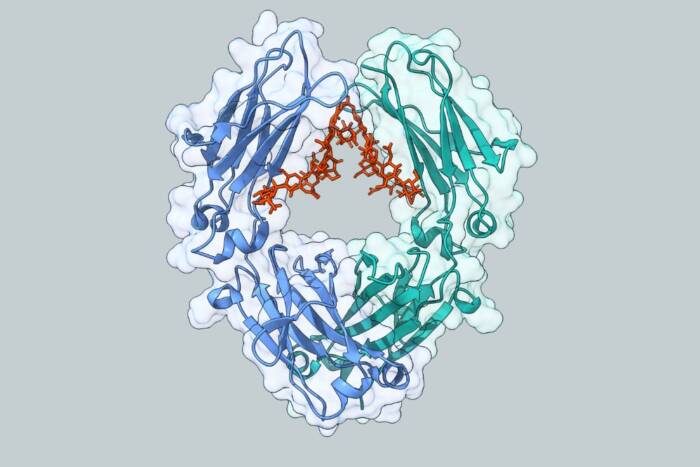
The multidisciplinary project grew out of research that McKinney initiated as a postdoctoral fellow in the laboratory of William R. Jacobs, Ph.D., a bacterial geneticist at the Albert Einstein College of Medicine and investigator with the Howard Hughes Medical Institute. Jacobs and McKinney, a 1994 Rockefeller University graduate, attacked the problem using bacterial genetics and animal infection models. Co-author David Russell, Ph.D., formerly of Washington University and now at Cornell University, focused on cell biology and enzymology. James Sacchettini, Ph.D., of Texas A & M University, solved the physical structure of ICL using the imaging technique known as X-ray crystallography.
In the Nature paper, the researchers demonstrated ICL’s link to persistence of the TB bug by infecting mice with two strains of M. tuberculosis-a “wild-type” strain containing the gene for ICL and a mutant strain that lacked the gene. During the early acute phase of infection, growth of the two strains was identical. Once infection entered the chronic phase, however, the mutant bacteria were largely eliminated by the immune system while the wild-type bacteria maintained peak loads in the animals. Not surprisingly, the mutant bacteria were less virulent. “We’re keeping mice that were infected with the mutant more than a year ago-they’re still alive and vigorous,” notes McKinney.
The research suggested that M. tuberculosis reprograms its carbon metabolism in response to the activation of certain immune cells-called macrophages-in the host. This idea was confirmed in subsequent experiments. Russell and colleagues showed that the mutant bacteria were robust in “resting” macrophages but were unable to survive in immune-activated macrophages. The requirement for ICL was correlated with the production of ICL by the bacteria, which was low in normal macrophages, high in activated macrophages. These results pointed to a link between the immune response of the host and the requirement for ICL. To confirm this link, McKinney and colleagues showed that when the mutant bacteria were injected into immune-deficient mice, their virulence was restored.
“This is a surprising finding-that the immune response of the host dictates the basic carbon metabolism of the pathogen,” McKinney says. “I don’t know of any precedent for this in the scientific literature.”
Click on the image below to see the animated version.
You will need version 4.0(opens in new window) or better of the Flash plug-in to see and hear this animation properly
 A separate paper, published by the same consortium of investigators in the August issue of Nature Structural Biology, reveals the three-dimensional structure of ICL. The work was led by structural biologist James Sacchettini, whose group also determined the structure of ICL in complex with specific inhibitory molecules that blocked the ability of the bacteria to use fatty acids as an energy source. Knowledge of the enzyme’s shape is guiding current efforts by collaborators at Glaxo Wellcome Medicines Research Centre in Stevenage, UK to identify drugs that will inhibit ICL’s function. These efforts, led by Ken Duncan, Ph.D., have already paid off handsomely, with several novel ICL inhibitors already identified and under study.
A separate paper, published by the same consortium of investigators in the August issue of Nature Structural Biology, reveals the three-dimensional structure of ICL. The work was led by structural biologist James Sacchettini, whose group also determined the structure of ICL in complex with specific inhibitory molecules that blocked the ability of the bacteria to use fatty acids as an energy source. Knowledge of the enzyme’s shape is guiding current efforts by collaborators at Glaxo Wellcome Medicines Research Centre in Stevenage, UK to identify drugs that will inhibit ICL’s function. These efforts, led by Ken Duncan, Ph.D., have already paid off handsomely, with several novel ICL inhibitors already identified and under study.
McKinney says the collaboration among the scientists at the various institutions was the only way the TB research could progress so quickly.
“None of us working alone could have gotten us where we are. Tuberculosis R&D is slow, difficult, and expensive, and it’s not a high priority in the U.S. because it mostly affects people in developing countries,” he says. “Drug companies are understandably reluctant to invest a lot of money up front in basic research on TB. But by attacking the problem simultaneously from many angles, we’ve made ICL an attractive target, and in just a few years we’ve moved from an idea to having leads that could form the basis of a full drug-discovery program. Considering that there have been no new drugs developed specifically for TB since the 1960s, that’s not too bad.”
TB looms as a major public health issue that is intensifying. About 16 million people worldwide have active TB, and an estimated eight million new cases occur each year. Nearly two billion individuals-a third of the global population-are believed to harbor the infection in its latent form. In the United States and elsewhere, the spread of the human immunodeficiency virus (HIV) and the compromised immune systems that result have greatly increased the number of people susceptible to the TB germ. A drug that targeted persistent bacteria could be used as an adjunct to conventional treatments to clear TB infections much more quickly. That could translate into millions of lives saved every year.
Support for the research reported in the Nature and Nature Structural Biology papers was provided by Glaxo Wellcome and a Program Project grant from the National Institutes of Health. In addition, the Helen Hay Whitney Foundation and the Howard Hughes Medical Institute supported the research reported in Nature, and the U.S. Department of Energy and the Robert A. Welch Foundation supported the research reported in Nature Structural Biology.